Input Struktur dan Manipulasi
1. Mengambar molekul dengan program ini relatif sederhana. Pilih unsur dari tabel periodik, kemudian di click dan ditarik dengan mouse. Dengan mouse kita dapat mengkontrol rotasi di sekitar ikatan, mengatur stereokimia molekul dan mengubah struktur.
2. Dengan mouse-controlled tools kita dapat melakukan seleksi, rotasi dan translasi serta mengubah ukuran struktur. Setting pada menu harus dimodifikasi terlebih dahulu untuk mengontrol operasi dari tools.
3. Untuk mengkonversi struktur 2D menjadi struktur 3D dapat dikerjakan dengan HyperChem’s model builder.
4. Penggunaan constraint terhadap struktur relatif mudah. Kita dapat melakukan constraint terhadap panjang ikatan, sudut ikatan, sudut torsi dan juga terhadap atom yang diinginkan.
Display Molekular (Molecular Display)
- Pilihan rendering : Ball-and-stick, fused CPK spheres dengan pilihan shading and highlighting. Juga vdW dots, cylinders dan overlapping spheres.
- Ribbon rendering untuk protein backbones, dengan pilihan sidechain display.
- 3D Isosurfaces atau 2D contour plots untuk: muatan total, kerapatan muatan, orbital molekul, kerapatan spin, potensial elektrostatik (ESP), ESP dipetakan pada 3D charge density surface.
- Pilihan isosurface rendering: wire mesh, Jorgensen-Salem, transparent dan solid surfaces, Gouraud shaded surface.
- Selama simulasi dapat ditampilkan rerata energi kinetik , energi potensial, energi total dan parameter molekul seperti panjang ikatan, sudut ikatan, dan sudut torsi.
- Animasi mode vibrasi dari spektra IR
Kimia Komputasi

Jenis Perhitungan
Terdapat beberapa tipe perhitungan, antara lain kalkulasi single point, optimisasi geometri, frekuensi vibrasi, pencarian keadaan transisi, simulasi dinamika molekular, simulasi dinamika Langevin dan simulasi Monte Carlo.
1. Perhitungan single point dapat digunakan untuk menentukan energi molekul dari struktur yang telah ditentukan (tanpa proses optimasi)
2. Perhitungan optimisasi geometri menggunakan algoritma minimisasi energi untuk mendapatkan struktur paling stabil. Tersedia 5 algoritma minimisasi.
3. Perhitungan frekuensi Vibrational dimaksudkan untuk mencari mode vibrasi normal dari suatu struktur teroptimisasi. Spektrum teroptimisasi dapat ditampilkan dan gerakan vibrasi yang berkaitan dengan transisi spesifik dapat dianimasikan.
4. Pencarian keadaan transisi dilakukan dengan menentukan struktur metastabil yang bersesuaian dengan keadaan transition menggunakan metode Eigenvector Following atau Synchronous Transit. Sifat-sifat molekulernya kemudian dapat dihitung. Dua metode untuk melokasikan keadaan transisi diimplementasikan di dalam HyperChem 5.
a) Metode Eigenvector Following sangat cocok digunakan untuk proses unimolekular atau setiap system molecular yang mode vibrasi naturalnya cenderung menuju ke suatu keadaan transition.
b) Metode Synchronous transit khususnya berguna jika reaktan dan produk sangat berbeda. Terdapat dua metodologi synchronous transit yang diimplementasikan di dalam HyperChem yaitu Linear synchronous Transit (LST) dan Quadratic Synchronous transit (QST).
5. Simulasi Molecular dynamics menghitung trajektori klasik untuk sistem molekular. Waktu pemanasan, keseimbangan dan pendinginan dapat diterapkan dalam simulasi ini dan juga dapat digunakan untuk proses-proses yang bergantung pada perubahan waktu. Simulasi dapat dilakukan pada energi konstan atau temperatur konstan.
6. Langevin dynamics simulations untuk memodelkan efek tumbukan pelarut tanpa memasukkan secara implicit molekul-molekul pelarut.
7. Simulasi Monte Carlo Metropolis berguna untuk mengeksplorasi konfigurasi yang mungkin dari suatu sistem dalam keadaan keseimbangan dan menentukan sifat sistem yang dinyatakan sebagai harga rata-rata untuk sekuruh system yang sudah berada dalam keadaan keseimbangan.

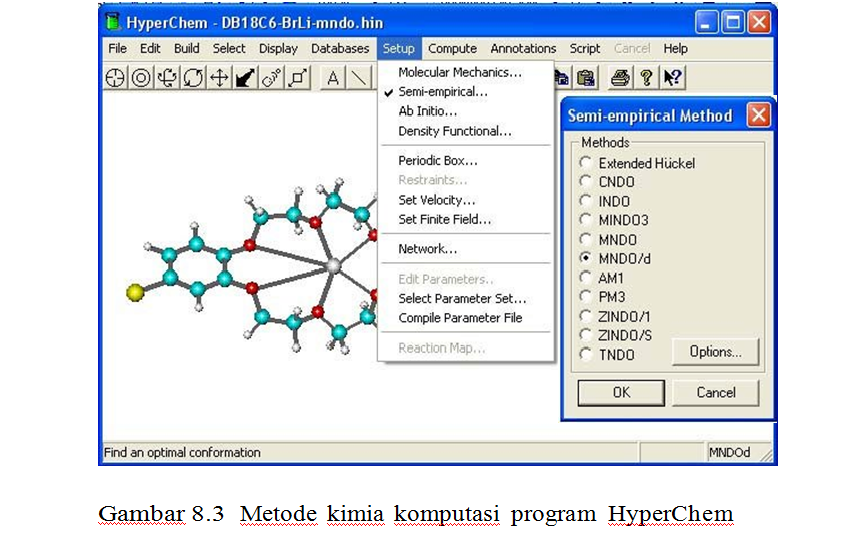
1. Metode mekanika kuantum ab initio.
• Tersedia pilihan beberapa himpunan basis di dalam program ini. Himpunan basis standar yang biasa digunakan antara lain STO-3G, 3-21G, 6-31G* dan 6-31G**.
• Fungsi-fungsi basis ekstra (s, p, d, sp, spd) dapat ditambahkan ke atom-atom individual atau ke sekelompok atom.
• Pengguna juga dapat mendefiniskan himpunan basisnya sendiri atau memodifikasi himpunan basis yang telah ada dengan menggunakan HyperChem‘s documented basis set file format.
• Tersedia pilihan beberapa himpunan basis di dalam program ini. Himpunan basis standar yang biasa digunakan antara lain STO-3G, 3-21G, 6-31G* dan 6-31G**.
• Fungsi-fungsi basis ekstra (s, p, d, sp, spd) dapat ditambahkan ke atom-atom individual atau ke sekelompok atom.
• Pengguna juga dapat mendefiniskan himpunan basisnya sendiri atau memodifikasi himpunan basis yang telah ada dengan menggunakan HyperChem‘s documented basis set file format.
2. Mekanika Kuantum Semiempirik.
• HyperChem menawarkan sepuluh metode molekular orbital semiempirik, dengan pilihan untuk senyawa dan senyawa-senyawa gugus utama, untuk senyawa transisi dan untuk simulasi spektra.
• Metode yang tersedia adalah Extended Huckel (oleh Hoffmann), CNDO dan INDO (oleh Pople dkk.), MINDO3, MNDO, MNDO/d dan AM1 (oleh Dewar dkk.) PM3 (oleh Stewart), ZINDO/1 dan ZINDO/S (oleh Zerner dkk.).
3. Mekanika Molekuler
HyperChem dapat digunakan secara mudah dalam menghasilkan struktur molekul 3D, dengan pilihan 4 metode mekanika molekular, teknik optimasi geometri untuk mendapatkan struktur stabil, dan teknik dinamika molekular untuk mendapatkan pencarian konformasi dan menginvestigasi perubahan struktur.
Penerapan metode mekanika molekular:
- Perhitungan energi konformasi relatif dari satu seri struktur analog (deret homolog).
- Reoptimasi peptida setelah ditentukan mutasi selektifnya
- Mendapatkan struktur yang mendekati realitas untuk perhitungan dengan metode kimia kuantum.
- Kebolehjadian terjadinya efek sterik pada zat antara reaktif.
Empat metode medan gaya (force field) memudahkan kita untuk mengeksplorasi stabilitas dan dinamika sistem molekular untuk senyawa yang mempunyai massa atom besar.
Untuk keperluan umum digunakan MM+, sedangkan untuk biomolekul dapat digunakan salah satu dari tiga metode medan gaya: AMBER, BIO+ dan OPLS.
MM+
• Sesuai untuk sebagian besar spesies non-biologi.
• Berdasarkan MM2 (1977) yang disusun oleh N.L. Allinger
• Menggunakan himpunan parameter 1991.
• Akan menjadi parameter default dalam kasus parameter MM2 tidak tersedia
AMBER
Empat metode medan gaya (force field) memudahkan kita untuk mengeksplorasi stabilitas dan dinamika sistem molekular untuk senyawa yang mempunyai massa atom besar.
Untuk keperluan umum digunakan MM+, sedangkan untuk biomolekul dapat digunakan salah satu dari tiga metode medan gaya: AMBER, BIO+ dan OPLS.
MM+
• Sesuai untuk sebagian besar spesies non-biologi.
• Berdasarkan MM2 (1977) yang disusun oleh N.L. Allinger
• Menggunakan himpunan parameter 1991.
• Akan menjadi parameter default dalam kasus parameter MM2 tidak tersedia
AMBER
- Sesuai untuk digunakan pada polipeptida dan asam nukleat dengan senua atom hidrogen diikutkan dalam perhitungan.
- Medan gaya AMBER force field disusun oleh Kollman.
- OPLS
- Didesain untuk perhitungan asam nukleat dan peptida.
- OPLS disusun oleh Jorgensen.
- Parameter interaksi tak berikatan dioptimasi dari perhitungan dengan pelarut termasuk di dalamnya.
BIO+
- Dikhususkan untuk perhitungan makromolekul.
- Medan gaya CHARMM disusun oleh Karplus.
- Disusun Primarily designed to explore macromolecules.
- Termasuk parameter CHARMM untuk perhitungan asam amino.
- OPLS disusun oleh Jorgensen.
- Parameter interaksi tak berikatan dioptimasi dari perhitungan dengan pelarut termasuk di dalamnya.
BIO+
- Dikhususkan untuk perhitungan makromolekul.
- Medan gaya CHARMM disusun oleh Karplus.
- Disusun Primarily designed to explore macromolecules.
- Termasuk parameter CHARMM untuk perhitungan asam amino.
Perhitungan dengan metode gabungan
HyperChem memungkinkan kita untuk menjalankan perhitungan kuantum terhadap sebagian dari sistem molekular, misalnya terhadap solut, sedangkan sisanya dihitung menggunakan metode klasik. Tehnik gabungan ini (QM/MM misalnya) dapat dijalankan untuk semua metode kuantum, hanya saja agak terbatas untuk pemakaian metode ab initio.
HyperChem memungkinkan kita untuk menjalankan perhitungan kuantum terhadap sebagian dari sistem molekular, misalnya terhadap solut, sedangkan sisanya dihitung menggunakan metode klasik. Tehnik gabungan ini (QM/MM misalnya) dapat dijalankan untuk semua metode kuantum, hanya saja agak terbatas untuk pemakaian metode ab initio.
Sumber : Prof. Dr. Harno Dwi Pranowo, M.Si (Kimia Komputasi)
1 comment:
kita juga punya nih artikel mengenai optimasi geometri, silahkan dikunjungi dan dibaca , berikut linknya
http://repository.gunadarma.ac.id/bitstream/123456789/3272/1/Kommit2004_kecerdasan_009.pdf
semoga bermanfaat
Post a Comment